Introduction
Sinus histiocytosis with massive lymphadenopathy (SHML), also known as Rosai-Dorfman disease, is a benign histiocytic disorder that was first described by Rosai and Dorfman in 1969.1 This condition is most commonly found in young adults, and its signs and symptoms include fever, high white blood cell count, and painless swelling of the cervical lymph nodes.1 Although SHML was classified for a long time as a non-Langerhans cell histiocytic disorder, it has recently been proposed that this disease is a new subtype denominated “group R” due to its unique features.2
Bone involvement has been reported in <10% of cases and primary osseous SHML is even less frequent.3 In these cases, bone lesions are usually multifocal and lytic and occur mainly in the cancellous tissue of long bones.3 Other diseases such as chronic recurrent multifocal osteomyelitis, metastasis, or Langerhans cell histiocytosis are also part of the differential diagnosis.4
The following is a case report of a 38-year-old man diagnosed with SHML who presented a lytic lesion in the medial cuneiform bone, which was treated with excision, curettage, and filling with bone graft.
Case Presentation
A 38-year-old man was evaluated by the dermatology service of a secondary care hospital in Palencia (Castilla y León, Spain) due to the presence of skin nodules on the thorax, scalp, and face. After performing a biopsy on one of the nodules, he was diagnosed with SHML. Six months after being diagnosed, the patient presented with mechanical pain in the right foot that increased with physical activity, so he attended a medical appointment at the same hospital. On physical examination, he presented pain in the inner side of the foot and the base of the first metatarsal, which increased when he was asked to use it for support. Otherwise, toe mobility was not restricted.
A non-enhanced x-ray was performed, revealing a lytic lesion in the medial cuneiform bone of the right foot, so CT scan and nuclear magnetic resonance imaging were requested. These tests confirmed the presence of a lytic lesion that destroyed the superomedial cortical bone of the medial cuneiform bone, which had a soft tissue component, a dimension of 21mmx19mmx15mm, and was adjacent to the base of the first metatarsal bone, although it had not infiltrated it (Figure 1).
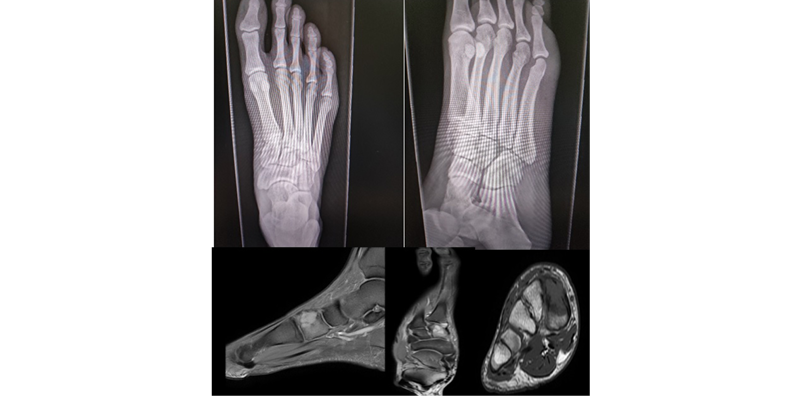
Figure 1. X-ray and magnetic resonance imaging of the right foot.
Source: Image obtained during the study.
Considering the anatomopathological, clinical and imaging (nuclear magnetic resonance) diagnosis of SHML, it was decided to remove the lytic lesion, without the need to perform a new biopsy. Under spinal anesthesia and ischemia of the lower limb, surgery was performed using a dorsal approach, including curettage of the lesion and filling with calcium phosphate cement HydraSet (Stryker®) (Figure 2).
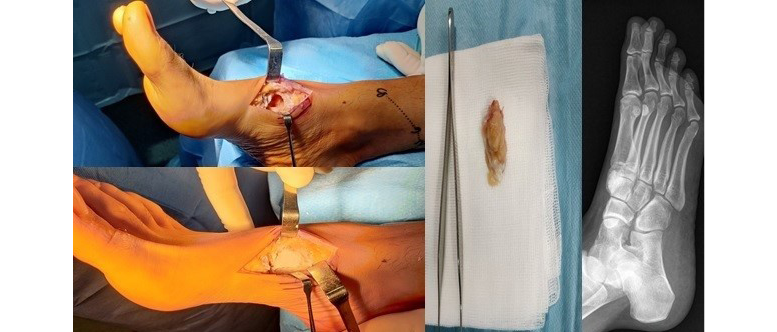
Figure 2. Surgical intervention performed on the right foot and postoperative X-ray.
Source: Image obtained during the study.
Subsequently, an anatomopathological study was performed on the excised tissue, which revealed a diffuse proliferative pattern consisting of large histiocytes with enlarged oval nuclei with a marked nucleolus and ample eosinophilic cytoplasm around them. Immunohistochemical testing identified positive staining of CD68 and S100 proteins, and negative staining of CD1a protein. Moreover, a moderate and polymorphonuclear lymphoplasmacytic inflammatory infiltrate was observed between histiocytes, indicating the presence of histiocyte proliferation, a finding consistent with SHML (Figure 3).
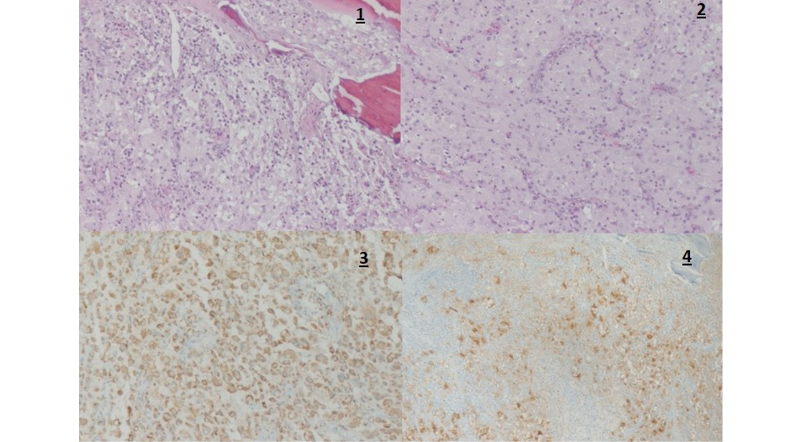
Figure 3. Anatomopathological study and immunohistochemical test of excised tissue from the right foot. 1) Cancellous bone surrounded by a diffuse proliferative pattern consisting mainly of enlarged histiocytes (hematoxylin and eosin stain, magnification x10); 2) Histiocytes showing oval nuclei with marked nucleoli and surrounding large eosinophilic cytoplasm (hematoxylin and eosin stain, magnification x20); 3) Positive immunohistochemical staining of S100 protein (magnification x20); 4) Positive immunohistochemical staining of CD68 protein (magnification x20).
Source: Image obtained during the study.
One year after surgery, the patient was asymptomatic and performed activities of daily living without restrictions, as well as impact sports without pain.
Discussion
SHML is a benign histiocytic disorder that was first described by Rosai and Dorfman in 1969.1 This disease is extremely rare and <1500 cases have been reported since that year.5 Patients with SHML usually present with painless swelling of the cervical lymph nodes.3
The pathogenesis of this disease is still unknown. However, recent studies have suggested that intermediate monocyte recruitment and alterations in their differentiation play an important role in its onset.3,4,6 It has also been reported that this condition may be the result of the presence of immunological diseases, as well as viral etiologies such as herpes simplex, parvovirus B19, or Epstein-Barr virus.6 On the other hand, the average age of people with SHML is 20 years, although cases of patients older than 70 years have been reported.5 This disease is more prevalent in men and in black people.5
Extranodal involvement is common and tends to be more frequent in the craniofacial massif. In turn, bone involvement is rare, occurring in 10% of cases, usually as a secondary process in patients with other sites affected by the disease. It has also been reported that primary bone involvement is extremely rare (2-8%).7 In addition, patients with SHML often present with pain in and swelling of the affected area, which can lead to respiratory or nervous system involvement in cases with lesions in the nostrils or intracranial lesions compressing peripheral nerves.
Regarding imaging tests, x-rays show lytic lesions with more or less well-defined edges, thinning or even rupture of the cortical bone and, sometimes, presence of shin splints.
Biopsy is the most important test for making the diagnosis of SHML. Findings related to this condition are characterized by the proliferation of numerous large histiocytes containing abundant eosinophilic cytoplasm and located within a mixed inflammatory infiltrate composed of plasma cells, lymphocytes, neutrophils, macrophages, and rare eosinophils. The distinctive feature of enlarged histiocytes is emperipolesis, i.e., the presence of lymphocyte phagocytosis with intracytoplasmic lymphocytes, plasma cells, or neutrophils.
Histiocytes in SHML are immunoreactive for CD68, CD163 and S100 proteins, and lack reactivity for CD1a protein. This immunohistochemical feature of SHML differentiates it from Langerhans cell histiocytosis, in which CD1a is immunohistochemically positive and histiocytes do not exhibit emperipolesis.8 In the present case, immunohistochemical testing yielded a positive result for CD68 and S100, and negative for CD1a, which is consistent with the typical findings of SHML.
This disease has a benign, proliferative and self-limited presentation, as well as an excellent prognosis. In general, patients with SHML do not need treatment, as spontaneous recovery occurs in almost 80% of cases.9 However, secondary involvement of vital organs and bones has been associated with an increased risk of death, mainly extreme extranodal involvement in children with central nervous system or kidney disorders, or respiratory tract involvement.10 In cases with systemic involvement, therapeutic options include excision of the lesion and treatment with corticosteroids, rituximab, and various chemotherapeutic agents.
The usual treatment in cases of SHML includes excision or curettage of the lesion, and filling with bone graft. Recurrence of the disease after surgery is very rare and may be caused by incomplete excision of the lesion or occur in patients with multiple organ involvement.9,10
Conclusions
SHML is a rare disease that affects not only the lymph nodes, but also other areas. Extranodal involvement is rare and bone involvement occurs in less than 10% of cases. This disease should be considered in the differential diagnosis of lytic bone lesions, especially when there are lesions in other parts of the body.
Ethical considerations
Informed consent was obtained from the patient for the preparation of this case report.
Conflicts of interest
None stated by the authors.
Funding
None stated by the authors.
Acknowledgments
None stated by the authors.
References
1.Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol. 1969;87(1):63-70.
2.Mosheimer BA, Oppl B, Zandieh S, Fillitz M, Keil F, Klaushofer K, et al. Bone Involvement in Rosai-Dorfman Disease (RDD): a Case Report and Systematic Literature Review. Curr Rheumatol Rep. 2017;19(5):29. https://doi.org/gd54.
3.Mansoori J, Fisher O, Akinyeye IO, Sobolevsky MA, Quinn RH. Primary Rosai-Dorfman Disease in 39-Year-Old Woman With Osseous Tibial Lesion Manifestion: A Case Report and Literature Review. Foot Ankle Orthop. 2021;6(4):24730114211060058. https://doi.org/jtwt.
4.Bruce-Brand C, Schneider JW, Schubert P. Rosai-Dorfman disease: an overview. J Clin Pathol. 2020;73(11):697-705. https://doi.org/gnts6w.
5.Abla O, Jacobsen E, Picarsic J, Krenova Z, Jaffe R, Emile JF, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131(26):2877-90. https://doi.org/gdsxww.
6.Mohammadi O, Zylberglait Lisigurski M, Mehra D, Pishdad R, Gulec S. Rosai-Dorfman Disease and Unusual Local Invasive Presentation. Cureus. 2020;12(3):e7328. https://doi.org/jtwv.
7.Ross AB, Davis KW, Buehler D, Chan BY. Primary Rosai-Dorfman Disease of Bone: A Report of Two Cases. Case Rep Radiol. 2019;2019:1720131. https://doi.org/jtww.
8.Izubuchi Y, Suzuki K, Imamura Y, Katayama H, Ohshima Y, Matsumine A. Primary Rosai-Dorfman disease of bone arising in the infantile ilium: A case report. Exp Ther Med. 2020;19(4):2983-8. https://doi.org/jtwx.
9.Vithran DTA, Wang JZ, Xiang F, Wen J, Xiao S, Tang WZ, et al. Osseous Rosai-Dorfman disease of tibia in children: A case report. World J Clin Cases. 2021;9(6):1416-23. https://doi.org/jtwz.
10.Warrier R, Chauhan A, Jewan Y, Bansal S, Craver R. Rosai-Dorfman disease with central nervous system involvement. Clin Adv Hematol Oncol. 2012;10(3):196-8.